
Note: for this installation, we now require multi-variate calculus, specifically partial differentiation.
10. More State Parameters
For this section, let’s consider the case of a homogeneous gas, whose state is parametrised by (P, V, N), where N is kept constant for now. Recall that the first law of thermodynamics says: dQ = dU + P·dV, where dQ is a small amount of heat added to the system and dU is a small change in the system’s internal energy. But we also know from definition that dQ/T = dS, which is a small change in the system’s entropy. This gives:
dU = T·dS – P·dV.
This suggests that if we’re looking at the state variable U, then it’s a good idea to re-parametrise it as U = U(S, V, N). The above equation then tells us:

[ For students who’re unfamiliar with partial differentiation, this means: if we keep the parameters V and N constant and differentiate U with respect to S, then the result is T. E.g. if 
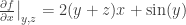
What if we wish to parametrise in terms of T, V and N? The best approach is then to define F = U – ST, the Helmholtz free energy. If we perturb the system by a little bit, then the change in F is:
dF = dU – S·dT – T·dS = –S·dT – P·dV.
If we parametrise F via F(T, V, N), then 
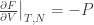
- It’s the maximum amount of useful work you can get out of a system at an isothermal setting. Imagine connecting the system to a heat source of temperature T (i.e. same temperature as the system) and letting it do work by changing P and V. This process is reversible. Let Q be the amount of heat the system extracts from the heat source.
- Since T = constant, change in entropy
.
- Change in internal energy is
.
- Thus the work done is
, which is the negation of the change in free energy. Hence one can envisage converting the free energy into work.
- Since T = constant, change in entropy
- It’s the amount of energy required to form the system when it’s connected to a heat source T. Without the heat source, the amount of energy required is clearly U. However, the heat source helps out by contributing ST.
To parametrise in terms of S, P and N, we let H = U+PV be the enthalpy of the system. Then dH = dU + P·dV + V·dP = T·dS + V·dP and we get:

This is the amount of energy required to create the system when it’s fixed at constant pressure P (called an isobaric setting): in addition to the internal energy U, the system has to do work in order to maintain its pressure. Since P is constant, the work done is PV.
Finally, as the reader might have guessed, for (T, P, N), we define the Gibbs free energy G = U + PV – ST. One obtains:

This is the amount of energy required to create the system in an isobaric and isothermal setting (i.e. its pressure is kept constant and at the same time, it’s connected to a constant heat source).

11. Intensive and Extensive Variables
Let’s summarise the large number of state parameters we have:
- pressure P,
- volume V,
- temperature T,
- entropy S,
- number of particles N,
- internal energy U,
- Helmholtz free energy F,
- Gibbs free energy G,
- enthalpy H.
Each of these state parameters can be classified from the following question: if we take the union of two identical systems, do we get the same parameter, or twice the parameter? If it’s the former, we say the parameter is intensive; for the latter, extensive. In this list, the only intensive state variables are P and T while the rest are extensive. Thus, for the internal energy U, we have:
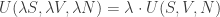
for any positive value of λ. We also get:
;
;
.
The second relation is particularly interesting: if T and P are kept constant, then G is linear in N! Hence we can write 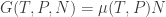

Example 9. Let’s go back to the problem of finding the entropy of an ideal gas. We had obtained:

To figure out the right t(N), we use the fact that S, V, N are extensive while P is intensive, i.e. 

Replacing 


We will leave it at that, since figuring out k is rather tricky business which requires quantum mechanics. The complete version of the equation, known as the Sackur-Tetrode equation, even depends on the mass of each particle!
12. Application of Thermodynamics: Introduction to Phase Transition
Phase transition occurs when the system experiences a sudden change in its physical properties. Typical examples that we see in everyday life include boiling / condensation and freezing / melting. The theory of thermodynamics allows us to model the behaviour of matter, if not quantitatively, then at least qualitatively.
The following is extracted and rephrased from David Tong’s notes on statistical physics, but any mistake most likely belongs to me. Most of the diagrams below are also stolen / modified from his notes.
Let’s write down van der Waal’s equation for a not-so-ideal gas/liquid:
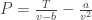
If we plot out the P–v graph (warning: not P–V graph) for various temperatures, we obtain the following:

Note that if V is close to b, then the material is in a liquid state since compressing it requires a disproportionately large amount of force. Conversely, if V is huge, then it is in gaseous state.
For temperatures below a critical limit 
How do we find the interval where vapour-liquid equilibrium occurs?
In order for vapour and liquid to coexist, the intensive parameters of both components must be equal. The temperature is already equal. Let the common pressure be P. The final parameter is the chemical potential μ (or the Gibbs free energy per particle), which is the Gibbs energy per particle since G(T, P, N) = μ(T, P) N. Thus we have:

Since our T and N are indeed fixed, we get 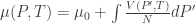

Hence, for (P, μ) of both the solid and liquid states to be equal, we need 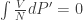
Now let the temperature increase until 

As one might imagine, this temperature is rather high (for water, it’s about 374°C). From van der Waal’s equation, this critical point can be determined via algebra, for it is where the P–v graph has an inflection point:

Solving gives 


And this concludes our very brief introduction to phase transition. Needless to say, we’ve barely scratched the surface of the applications of thermodynamics, but we hope the reader’s interest is sufficiently piqued to seek out other more definitive sources.
